En el transcurso de una reunión científica en Alta (California), en 1984, un grupo de investigadores abordó el debate sobre la conveniencia de poner en marcha un ambicioso programa. Un proyecto encaminado a la detección de aquellas mutaciones génicas causantes de enfermedades. Sería dos ańos después, durante un congreso en Santa Fe (California), auspiciado por el Ministerio de Energía (DOE), cuando formalmente se propuso el “Human Genome Project”: como medio para afrontar sistemáticamente la evaluación del efecto de las radiaciones sobre el material hereditario. El proyecto se presentaba ambicioso, tanto en su presupuesto económico como en su duración. Para James Watson, Premio Nobel junto con Francis Crick por el descubrimiento de la estructura de la “doble” hélice del ADN e impulsor del proyecto: “Aunque el costo global de la secuenciación total del ADN humano será inferior en un orden de magnitud al de enviar al hombre a la luna, las repercusiones serán mucho mayores”. Se trataba, en fin, de descifrar el genoma humano para poder comprender y erradicar el cáncer, así como otras enfermedades de determinación génica. Un proyecto de esta envergadura sólo podía plantearse partiendo de los grandes avances en Biología Molecular y en las Ciencias de la Computación. Por lo que se refiere a la Biología ha sido determinante el gran desarrollo, a partir de la década de los ochenta, de la metodología del ADN recombinante con todas sus técnicas asociadas: vectores de clonación, enzimas de restricción, secuenciación genética inversa, reacción en cadena de la polimerasa,… En cuanto a la Bioinformática, no sólo está permitiendo la secuenciación de genomas, sino la detallada comparación de los mismos. Más allá de las aplicaciones terapéuticas que pudieran derivarse del desciframiento de nuestro genoma, en el trasfondo del proyecto subyace una motivación no menos ambiciosa: profundizar en el enigma insoldable sobre nuestro origen. En este sentido, son esclarecedoras las palabras de Jacques Monod (1977), Premio Nobel por sus investigaciones sobre la expresión de los genes: “la ambición última de la ciencia entera es fundamentalmente dilucidar la relación del hombre con el universo… a la Biología le corresponde un lugar central, por ser la disciplina que intenta ir más directamente al centro de los problemas que se deben haber resuelto antes de poder proponer el de la «naturaleza humana»”. Y no menos significativas las de Francis Crack: “un hombre honesto que estuviera provisto de todo el saber que esté hoy a su alcance, debería afirmar que el origen de la vida parece provenir de un milagro… Sólo un milagro puede definir las condiciones que sería preciso reunir para el establecimiento de la vida”. Y es el que el proyecto, según Walter Gilbert (Premio Nobel por el desarrollo de una de las técnicas de secuenciación), “traerá consigo un cambio en la concepción filosófica sobre nosotros mismos”. El Proyecto Genoma Humano (PGH, en su acrónimo) fue refrendado por el Congreso Norteamericano en 1988, tras recibir la aprobación de la Academia de las Ciencias, el Instituto Nacional de la Salud y el Gobierno. Y se puso en marcha en 1990 en Estados Unidos. Posteriormente entraron a participar Gran Bretańa, Francia, Alemania, Japón y China. Esta internacionalización exigió la creación del “Human Genome Organization” (HUGO): un órgano rector y coordinador presidido, inicialmente, por el genetista Victor McKusic. El planteamiento original (1988) marcaba ocho objetivos, encaminados a habilitar soluciones mediante técnicas de terapia génica u otras aplicaciones biotecnológicas: - Descifrar todo el genoma humano. Es lo que se conoce como “genómica estructural”: żqué información contiene el genoma?
- Desarrollar una tecnología eficiente para la secuenciación de todo el genoma.
- Identificar las variaciones alélicas.
- Interpretar las funciones. Es lo que se conoce como “genómica funcional”: żqué codifican los genes?, żqué genes, en qué orden, dónde y cuándo se expresan en relación con el desarrollo?
- Descifrar y analizar las secuencias de “organismos modelos”. Concretamente: una levadura (Saccharomyces cerevisiae), un nemátodo (Caenorhabditis elegans), la mosca de la fruta (Drosophila melanogaster) y el ratón común (Mus musculus).
- Examinar las implicaciones éticas, legales y sociales de la investigación genómica. El programa internacional ELSI (“Ethics, Legal and Social Implications”).
- Desarrollar herramientas de bioinformática y estrategias de computación para el uso de los datos de genes y secuencias.
- Adiestrar científicos para la investigación y análisis de los genomas.
Por la parte estadounidense han corrido con la financiación y la coordinación: NHGRI (“National Human Genome Research Institute”), NIH (“National Institute of Health”) y DOE (“Department of Energy”). Y por la británica: el “Wellcome Trust”. Inicialmente en el proyecto se implicaron dieciséis centros de investigación de Estados Unidos, además de numerosos departamentos de los otros países partícipes. En base a los ocho objetivos seńalados se marcaron dos etapas: la primera de ellas, referida a la genómica estructural, habría de finalizar concluyendo el 2001 con la realización de un “borrador” del genoma. Sin embargo, esta primera fase culminó antes de lo previsto: en el 2000 se anunciaba ese “working draft” del genoma. En esta acelerada consecución de objetivos ha resultado relevante el desarrollo de un amplio programa de investigación pública y, paralelamente, otro privado: dirigido por el Dr. Craig Venter, del grupo “Celera Genomics”. No menos importante es el carácter altruista que, hasta la fecha, ha caracterizado el desarrollo del programa. Así por ejemplo, todos los hallazgos se han ido publicando en Internet en tiempo real. Si bien el Proyecto Genoma Humano se inició en 1990, ya desde los ańos setenta y ochenta se venían descifrando genomas de muy diferentes organismos. Entre 1976 y 2004: seis virus, once bacterias, un hongo, dos plantas, dos invertebrados, dos vertebrados, así como el ADN de las mitocondrias humanas. En la Tabla-I se exponen algunos ejemplos. Inicialmente el PGH se presupuestó en 3.000 millones de $ USD, estimándose su duración en quince ańos. Ahora bien, las innovaciones tecnológicas han posibilitado un considerable recorte en costes y tiempo. La primera fase concluyó en trece ańos, invirtiéndose 2.600 millones de $ USD. Reseńable es la creciente relevancia que ha ido tomando la investigación privada. En 1993 los fondos públicos ascendían a 170 millones de $ USD, frente a los 80 invertidos por compańías biotecnológicas privadas. En mayo de 1998 la empresa TIGR formó un consorcio con Perkin-Elmer, un fabricante de secuenciadotes automáticos, con el fin de abaratar y abreviar el proyecto público. De hecho, esta carrera competitiva está reduciendo costes. A modo de ejemplo, el aislamiento y secuenciación del gen responsable de la fibrosis quística costó 30 millones de $ USD, actualmente sólo habría supuesto 200.000. 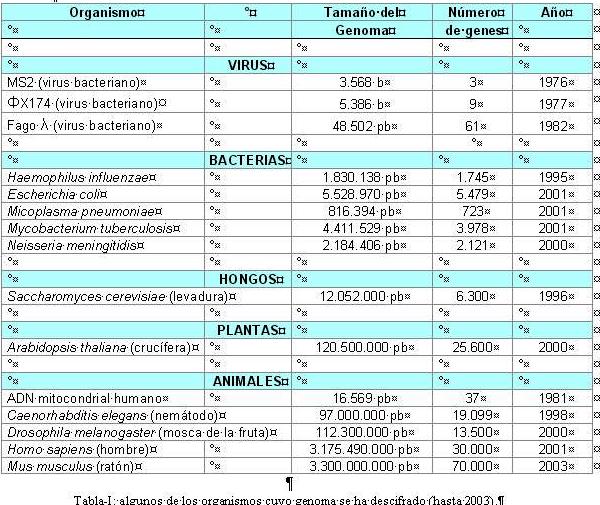 Dos técnicas para un mismo proyecto Desde los comienzos de su andadura el Proyecto Genoma Humano ha contado con un aliciente extra. Nos referimos al empleo de dos técnicas, paralelamente, en la secuenciación del genoma. Evidentemente, cuanto más amplio es el enfoque de estudio sobre un hecho: más fino resulta el análisis final y más fiables las conclusiones. Ambas metodologías fueron decisión de los principales investigadores, de su -podríamos decir- propia idiosincrasia o metodología de trabajo. La primera de las estrategias fue la propuesta por James Watson y Francis Collins, entre otros, contando con la mayor parte de la financiación pública. Esta metodología resulta la más compleja, pues se enfoca a un conocimiento más exhaustivo del genoma. Es, por ende, la de uso más generalizado. Si bien más adelante detallaremos su método, podemos resumirlo como sigue. Básicamente consiste en secuenciar el genoma completo, cromosoma a cromosoma: de un extremo al otro. Podríamos hablar de “fragmentación total”, que es la que explicaremos, por su mayor complejidad, con mayor detalle. Craig Venter y otros, bajo la financiación de “Celera Genomics” y otras compańías biotecnológicas privadas, emplearían una segunda técnica, más práctica. Consistiría en la secuenciación de genes expresados en las células diferenciadas en las que se encuentran activos, partiendo de los ARNm resultantes de su traducción. Fragmentación total La técnica propuesta por Watson, Collins et al, podría ejemplificarse con el clásico “corta y pega”, a no ser por la magnitud de la secuencia a analizar. Podría especificarse en los siguientes pasos. - Aislamiento del ADN, partiendo de núcleos de células, usualmente, en cultivo.
- Escisión del genoma en miles de fragmentos manejables. E inserción de los mismos en los llamados “vectores de clonación” (plásmidos, por ejemplo).
- Clonar dichos fragmentos en microorganismos, lo que posibilita la conservación “aislada” de secuencias de ADN perfectamente “ubicadas” (aunque aun no descifradas). Es lo que se conoce como “librerías genómicas”. Es decir, insertar los vectores portadores de fragmentos en microorganismos, manteniendo estos en cultivo.
- Aislamiento de los mencionados fragmentos, a partir de las “librerías”.
- Localizar las regiones contiguas (los puntos de cohesión) entre los fragmentos, de forma que, analizando detalladamente la secuencia se determinen esos “cóntigos” o fragmentos contiguos.
- Secuenciación de cada fragmento y ensamblado completo de todas las secuencias dentro de sus “cóntigos”.
Fragmentación y clonación La fragmentación del ADN sería imposible sin el empleo de las endonucleasas de restricción, unas enzimas de Escherichia coli, descubiertas por Werner Arber en los ańos setenta, implicadas en su defensa contra la infección por bacteriófagos. Las endonucleasas son capaces de cortar la secuencia de ADN, independientemente de su origen, en lugares específicos. Esta especificad hace de estas enzimas unas herramientas sumamente precisas. Un ejemplo es la enzima EcoRI, que reconoce y corta la siguiente secuencia de seis pares de nucleótidos en el ADN de cualquier organismo: 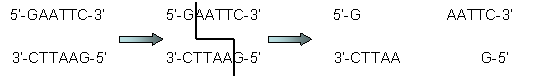 Se generan así un par de extremos cohesivos, que pueden unirse entre sí o con cualquier otro extremo con una secuencia complementaria: es decir, con un fragmento cortado por la misma enzima. Se denominan “palíndromos” a esas secuencias diana que son reconocidas por las endonucleasas de restricción. El término hace referencia a que ambas cadenas de ADN tiene comparten la misma secuencia nucleotídica en una orientación antiparalela, conforme a la corresponsabilidad de bases: Guanina con Citosina, Adenina con Timina. En general, la mayoría de estas enzimas reconocen palíndromos específicos. Fragmentado pues el ADN en trozos, que puedan contener uno o varios genes, el siguiente paso sería la clonación de los mismos. Para ello, se insertarán en moléculas con capacidad de autorreplicación: plásmidos, cósmidos, cromosomas artificiales de levadura (YACs), cromosomas artificiales de bacteria (BACs),… Cada uno de estos vectores aceptan secuencias de distinta longitud: los cósmidos unas 40 kb, los YACs entre 100 y 2.000 kb, por ejemplo. Los plásmidos son pequeńos anillos de ADN (de doble cadena) extracromosómico, presentes en microorganismos tales como las bacterias, que pueden utilizarse como vectores. Para la inserción de ADN foráneo en los plásmidos, se recurre a las comentadas endonucleasas de restricción. Por ejemplo, puede cortarse el ADN vector (el del plásmido) y el ADN donante (el humano, pongamos por caso) con la enzima EcoRI, generándose unos extremos cohesivos que podrán unirse entre sí constituyendo un vector recombinante. Finalmente, el plásmido modificado será insertado en una bacteria receptora, en orden a su clonación (Fig.-1). 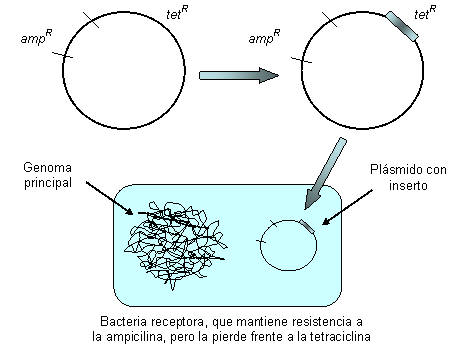 El procedimiento parece sencillo, pero a priori plantea una duda razonable. żCómo se puede tener certeza de que el ADN donante se ha insertado eficientemente en el plásmido vector? Para la resolución de este problema, la Biología Molecular se aprovecha de una propiedad de los plásmidos: el hecho de que porten genes que les confieren resistencia a antibióticos. Así por ejemplo, el plásmido pBR322 (un anillo de ADN de doble hélice, de 4.600 pares de bases) porta dos genes de resistencia a dos antibióticos: el tetR, frente a la tetraciclina; el ampR, frente a la ampicilina. Ambos genes contienen secuencias diana de corte específico para determinadas endonucleasas de restricción: HindIII, BamH1 y SalI para tetR; PstI, PouI, EcoR1, AvaI, PouII y ClaI para ampR. Al tratar el plásmido pBR322 con la enzima BamH1 resultará cortado el gen tetR, con lo que el anillo quedará abierto y receptivo para la inserción del ADN donante. Consecuentemente, el plásmido perderá su resistencia a la tetraciclina. Una vez mezclado el plásmido “abierto” y el ADN foráneo, ambos tratados con la BamH1, es de esperar que este último fragmento se inserte en el primero: cerrándose de nuevo el anillo del primero. Para cerciorarse de una eficiente inserción, se introducirán estos plásmidos en bacterias para su multicultivo. Se seleccionarán las colonias bacterianas que muestren resistencia a la ampicilina y la hayan perdido frente a la tetraciclina. Una bacteria transformada con una sola molécula de ADN recombinante comenzará a dividirse exponencialmente, en un medio sólido, dando lugar a una colonia con millones de células hijas, todas ellas portando ese fragmento de ADN foráneo inserto en un plásmido. De este modo, se van obteniendo colonias transformadas de bacterias, que constituirán clones portadores de un determinado plásmido con un mismo inserto de ADN donante. Cada colonia sólo habrá de contener una única molécula recombinante del fragmento de genoma que se desea estudiar. Esto no significa, sin embargo, limitaciones extremas en los fragmentos de ADN foráneo a insertar en los plásmidos, ni tampoco en el tipo de vectores de clonación. Se pueden insertar secuencias nucleotídicas de mayor o menor longitud. No obstante, en orden a afinar lo más posible el desciframiento se ha procedido a ordenar los pequeńos fragmentos, pertenecientes a una común región mayor, entre sí: y así, sucesivamente, en orden ascendente. Se obtendrá así una inmensa colección de clones, para cada fragmento del ADN original seccionado (sobre un total de 3.175.490.000 pares de bases y 30.000 genes, en el caso del ser humano): lo que se conoce como una “librería genómica”. Librerías genómicas Debido a los distintos tipos de vectores empleados, así como al tamańo de las secuencias insertadas -como más arriba comentábamos-, para el “Human Genome Project” se han constituido varias de esas librerías genómicas. Una simplificación del trabajo, con vistas a facilitar la reconstrucción (desfragmentación) del ADN previamente fragmentado, ha llevado a partir de cada uno de los cuarenta y seis cromosomas aislados: el trabajo se planteó cromosoma a cromosoma. Así, una librería genómica habrá de albergar miles o millones de fragmentos del ADN a estudiar. Pero, żcuál sería su magnitud? Por lo general, el número de clones será, cuando menos, el doble de la suma de los tamańos medios de las secuencias insertadas, una vez fragmentado el genoma. Por ejemplo, un genoma de 3.000 Mb tendría cabida en 3.000 cultivos de levadura (una región de 1 Mb/cultivo), o bien en unos 210.000 plásmidos (una región de 15 kb/plásmido). Y aún así, el número de cultivos habría de ser el doble, puesto que las endonucleasas de restricción reconocen unas específicas secuencias dianas y, por tanto: i) pueden quedar zonas de corte fuera de su acción; ii) pueden cortar segmentos de ADN, más o menos largos, que representen la misma región del genoma (con segmentos comunes y otros que solapen con regiones colindantes). żCómo ordenar? Llegados a este punto, nos encontraríamos en una situación que podríamos calificar de “sin orden ni concierto”. żCómo ordenar, cómo reconstituir? En el proceso de enlazado hay que buscar regiones solapantes entre los fragmentos: secuencias comunes en los insertos clonados. Así se irán obteniendo cóntigos (“contigs”): conjuntos de fragmentos de un genoma que se han clonado por separado, pero que son contiguos y que están parcialmente solapados. En la elaboración de los mapas físicos del genoma, se irán ensamblando dichos “contigs”. Se utilizan marcadores moleculares. Por la técnica de la PCR (Reacción en Cadena de la Polimerasa), desarrollada por el Premio Nobel Kary B. Mullis, se analizarán detalles de secuencias. Los marcadores indican las posiciones de cortas secuencias (Fig.-2), únicas y comunes a todos los fragmentos indicados; de forma que así se pueden conocer la posición y el orden de éstos. Hay un tipo de marcadores particularmente eficaces, por su alta resolución y rapidez de ejecución: los STS (“Sequence Tagged Sites”: lugares etiquetados por su secuencia). Se trata de secuencias cortas de ADN, de entre 100 y 1.000 pb conocidas, fácilmente reconocibles y presentes, únicamente, en un lugar de un cromosoma o del genoma. Los STS pueden detectarse en distintos fragmentos que tengan extremos solapantes, pero que se hayan conservado en clones separados. Una vez que un investigador descifra un STS, cualquier otro puede obtener su secuencia fabricando in vitro los cebadores correspondientes a sus extremos, y amplificando la STS mediante la mencionada PCR. Los STS constituirían una suerte de balizas en la elaboración de los mapas físicos. Entre los objetivos principales del proyecto figuraba la elaboración de mapas físicos con alrededor de 30.000 de esas balizas (STS), quedando los marcadores separados entre sí por unas 100 kb. Objetivo ya cumplido, gracias precisamente al empleo de los STS: se han obtenido mapas de cóntigos conforme al contenido de marcadores de los clones solapantes. Secuenciación En esta fase final del proceso es donde técnicas como la nanotecnología, la bioinformática y la biología computacional han resultado de inestimable ayuda. Los avances en las nuevas estrategias de secuenciación automática han posibilitado secuenciar un fragmento de 700 bases en pocos minutos. Además, un secuenciador automático puede realizar cerca de 100 análisis simultáneos. Es conocido el famoso laboratorio francés “Génethon”, provisto de varios robots especializados en procesar y analizar las muestras. Se puede secuenciar un fragmento de ADN amplificado por PCR, o directamente un inserto en un plásmido. 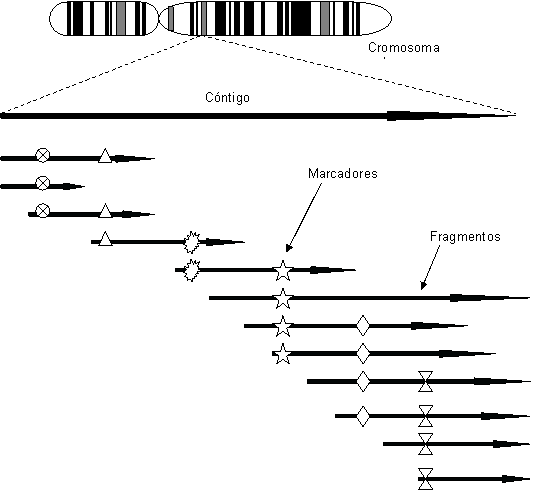 Existen varios métodos de secuenciación. De los siguientes, los tres primeros son los básicos, en tanto que el resto corresponden a nuevas metodologías aún en desarrollo. - Método químico de Maxam y Gilbert. Actualmente en desuso.
- Método enzimático de Sanger, también conocido como de “terminación de cadena”, de “secuenciación automática”, o de los didesoxinucleótidos (ddNTP). Más adelante nos detendremos en explicarlo con más detalle.
- Microscopía de barrido (“scanning”) de efecto túnel (STM). Próxima a la muestra de ADN se mantiene una sonda, mediante un control basado en la detección de una ínfima corriente eléctrica inducida por el “efecto túnel” entre la punta de la sonda y el ADN. Los movimientos en vertical de la sonda a lo largo de la muestra se irán midiendo y registrando, generándose una imagen de la superficie del ADN. Se han obtenido imágenes del esqueleto desoxirribosa-ácido ortofosfórico, pero aún no de las bases nitrogenadas. De llegar a desarrollarse satisfactoriamente, este método podría secuenciar 1 Mb cada día.
- Microscopía de fuerza atómica (AFM). Método similar al anterior, donde el control de la sonda se debe a la medición de las fuerzas de van der Waals entre aquella y la muestra.
- Secuenciación por hibridación en chips con oligonucleótidos. Esta técnica bioinformática se basa en la síntesis de distintas sondas de oligonecleótidos, que luego se unirán en disposiciones ordenadas (“arrays”) sobre un “chip”. Este se probará frente a una muestra de ADN fluoromarcado: el patrón y cantidad de fluorescencia emitada informará sobre la secuencia del ácido desoxirriblonucleico. Una técnica donde la nanotecnología está logrando llamativos avances. La compańía “Affimetrix”, por ejemplo, ha conseguido resecuenciar las 16’5 kb del ADN mitocondrial humano, mediante un “biochip” de 135.000 oligonucleótidos (de unas 20 bases cada uno). En el 2003 el Centro Nacional de Investigaciones Oncológicas, dirigido por el Dr. Mariano Barbacid, presentó el primer “biochip” espańol.
Por lo que respecta al método de Sanger, habitualmente se utiliza una síntesis de secuencias complementarias mediada por una polimerasa, y se emplea la molécula a secuenciar como molde: molde que irá incorporando nucleótidos en la nueva cadena. En la mezcla de reacción se ańadirán los cuatro dNTPs (deoxinucleótidos trifosfatos de A, T, G y C), que tienen el grupo 3’-OH (enlace fosfodiester entre 3’-OH y 5’-OH del siguiente dNTP), así como los cuatro ddNTPs (dideoxinucleótido trifosfatos de A, T, G y C), caracterizados por carecer de la terminación 3’-OH (en cualquiera de las bases). Esta modificación presente en los ddNTPs impide la unión de nuevos nucleótidos, en su incorporación a la nueva molécula que se va sintetizando. Un fluorcromo específico marca cada uno de los ddNTPs, generando una seńal de color diferente. Al incorporar un ddNTP marcado se interrumpe la síntesis de la molécula; al tiempo, se incorporan otros dNTPs no marcados. Así, la menor concentración de ddNTPs (marcados) frente a dNTPs (no marcados), hace que al final de la síntesis sólo se produzcan incorporaciones ocasionales. Transcurrido un cierto tiempo, de la reacción de síntesis resultarán numerosas moléculas de diversa longitud, que finalizarán en la posición ocupada por una base característicamente marcada por una seńal fluorescente (A, T, C o G): Fig.-3.  Fig.-3: Lectura de síntesis resultante con marcaje fluorcromo de ddNTPs . Seguidamente se separarán, en función de su tamańo, los productos de la reacción de síntesis mediante una electroforesis. La separación se efectúa a tiempo real mediante un lector láser. Los secuenciadores automáticos emplean programas altamente sofisticados, que integran todos los resultados para concluir una secuencia gráfica. Mapas genómicos Entre los principales objetivos del PGH figura la creación de una serie de mapas, cada vez más finos de cada cromosoma humano. Trazar un mapa supone dividir los cromosomas en fragmentos menores, que se puedan amplificar, caracterizar y ordenar. El segundo objetivo, una vez obtenido un mapa, sería determinar la secuencia de ADN de cada uno de los fragmentos ordenados. En último término se trata de encontrar todos los genes en la secuencia de ADN. Se distinguen tres tipos de mapas: - Mapas de ligamiento genético. Muestran la localización relativa de marcadores específicos de ADN a lo largo del cromosoma. Los marcadores son regiones de ADN (codificantes o no) cuya forma o patrón de herencia puede seguirse. Dos marcadores localizados uno junto a otro en el mismo cromosoma, tienden a pasar juntos de padres a hijos. Los marcadores deben ser polimórficos (color de ojos, grupos sanguíneos, susceptibilidad a una enfermedad,…).
- Mapas físicos. Según el nivel de resolución, pueden obtenerse diferentes mapas físicos:
1. Mapa cromosómico. Se trata de un mapa físico de baja resolución, basado en el patrón de bandas distintivo que presentan los cromosomas teńidos cuando se observan al microscopio óptico. 2. Mapa de ADNc (ADN clínico). Muestra la localización de los genes (exones) en el mapa cromosómico. El ADNc identifica las partes del genoma con mayor importancia biomédica. 3. Mapa de “contings”. Por “cotings” (cóntigos) se entienden fragmentos de ADN ordenado que forman un bloque continuo. Este tipo de mapa físico de alta resolución construye una colección de fragmentos pequeńos (“contings”), de manera que se superponen estos fragmentos con un orden conocido y que se corresponden a un segmento completo de un cromosoma. 4. Mapa de restricción. De menor resolución que los anteriores, se construyen a partir de un cromosoma individual. El cromosoma se corta con enzimas de restricción, resultando fragmentos grandes que luego se ordenan y subdividen en fragmentos más pequeńos, que a su vez se reordenan. El mapa resultante muestra el orden de los sitios de restricción, así como la distancia que existe entre ellos. - Secuenciación del ADN. El mapa físico definitivo del genoma humano será la secuencia de ADN completa, donde se determinen todos los pares de bases de cada uno de los cromosomas.
En la secuenciación y ordenación, no sólo se puede recurrir a diferentes técnicas, sino que puede recurrirse a diversos vectores de clonación. En este sentido, la cartografía genómica de “contings” suele aprovechar la diversa capacidad de insertos para cada clase de vector con capacidad de autorreplicación. Las posibilidades son varias y no excluyentes. Veamos un ejemplo. Un primer paso sería la realización de mapas físicos de baja resolución. Para ello se partiría de clones solapados de aquellos insertos con mayor longitud: los YACs (entre 100 y 2.000 kb). Seguidamente se amplificaría la resolución de los mapas, subclonando en cósmidos (con capacidad para insertos de 40 kb) fragmentos aleatorios de aquellos mayores en YACs. Como última fase, se fragmentarían aleatoriamente los insertos de cósmidos clonados, para subclonarlos en vectores de menor capacidad como los plásmidos (15 kb de capacidad de inserto/plásmido) o virus como el fago M13 (400 pb de capacidad de inserto/fago). Aunque esta secuenciación aleatoria (“shotgun”) garantiza una alta fiabilidad, al secuenciar entre 8 y 10 veces un mismo segmento, sigue ofreciendo una tasa de errores considerable: entre el 0’02 y el 0’2 %. Y es que los YACs son vectores inestables, que frecuentemente se comportan como quimeras por su no absoluta fidelidad. Este modelo de secuenciación requiere disponer, previamente, de una cartografía aceptable. En 1996 Venter et al desarrollaron una estrategia alternativa, recurriendo a cromosomas artificiales de bacterias (BACs), para paliar las dificultades surgidas con los de levadura (YACs). Los BACs permiten insertos menores (100-350 kb), pero ofrecen una mayor fidelidad. Aunque económicamente ventajosa, la estrategia BAC no está tampoco exenta de polémicas: centralizaría el PGH, pues obligaría al intercambio de placas con clones. Esta novedosa técnica puede explicarse como sigue: - Creación de una genoteca humana de BACs, con un tamańo medio de insertos de 150 kb y alrededor de 300.000 clones.
- Para cada extremo del inserto de cada clon se secuencian unas 500 bases. Así, las 600.000 secuencias (llamadas STCs, o “conectores etiquetados por su secuencia”) de todos los extremos se reparten a razón de una cada 5 kb de genoma: constituyendo el 10 % de la secuencia genómica. Estas secuencias STC se hacen públicas, en tiempo real, en Internet.
- Los STCs posibilitan que, en términos medios, cada BAC pueda conectarse con otros 30. Un inserto medio de 150 kb dividido por 5 kb, está representado en 30 BACs.
- El paso siguiente sería tomar la “huella dactilar” (un patrón compartido de dianas para endonucleasas de restricción) de cada clon BAC.
- Para identificar los clones solapados se secuenciará un clon BAC y se comparará con la base de datos STCs: es el denominado “BAC semilla”.
- Secuenciación de los dos clones BAC que muestren consistencia interna entre las huellas dactilares y el solapamiento mínimo en ambos extremos en relación al “BAC semilla”.
- La repetición de este proceso, a ambos lados del “BAC semilla”, permitiría secuenciar todo el genoma humano a partir de sólo 20.000 clones BAC.
Resultados del Proyecto Genoma Humano Cumpliendo las perspectivas del PGH en 1990, en los primeros ańos del 2000 se han hecho públicos los genomas de varios organismos modelos muy diferentes (Tabla-I). Ya se conocen los genomas completos de más de cien bacterias y seis eucariotas. Se trabaja en el genoma de alrededor de mil especies de todos los phylums. Por lo que se refiere al ser humano, cuyo genoma es el objetivo central del proyecto, el primer éxito lo constituyó la publicación, a finales de 1999 (revista Nature), de la secuencia completa de del más pequeńo de nuestros cromosomas: el 22, con 33 Mb y la información de unos 650 genes. La misma revista publicaría, el 8 de mayo de 2000, la secuencia de un segundo cromosoma especialmente relevante, por su implicación en varias mutaciones (Síndrome de Down, por ejemplo): el 21. Y el 6 de junio de 2000, en la Casa Blanca y ante el Presidente Bill Clinton, se presentó un avanzadísimo -aunque incompleto- “working draft”, el borrador del genoma humano, por parte de Francis Collins, en nombre del consorcio público, y Craig Venter, por la privada Celera Genomics. Coincidiendo con el quincuagenario aniversario del descubrimiento de la estructura en doble hélice del ADN, el 13 de abril de 2003 Francis Collins hacía pública la culminación de la primera fase del PGH, aquella referida a la genómica estructural: completándose así el borrador, la secuenciación podía darse por concluida. Ha habido que afrontar un problema paralelo, de gestión e índole más práctica: la publicación de una ingente cantidad de datos, su disponibilidad en tiempo real y su libre accesibilidad. Obviamente, la publicación de secuencias de ADN en revistas científicas no ofrece, precisamente, facilidad alguna a los investigadores interesados en su consulta. Además, el volumen de datos se ha ido duplicando cada ańo. Desde sus inicios, los resultados del PGH se han ido publicando en varias páginas web para su consulta en Internet. Dos son las principales bases de datos genómicos: - El Consorcio Internacional de Bases de Datos de Secuencias, constituido por el Genbank, el Banco de datos de ADN de Japón (DDBJ) y el del EMBL. Estas tres genotecas intercambian información sobre secuencias. A su vez, se interconexionan con otras bases, como la del Centro Nacional de Biotecnología en Madrid.
- La Genome Data Base (GDB), más restringida a la cartografía y metodología en sí.
Para aquellas enfermedades cuyo origen genético ya ha sido determinado, es posible localizar los mapas genéticos e incluso las secuencias. Ello es posible gracias al vínculo entre la base de datos bibliográfica MEDLINE con la OMIM (“Herencia mendeliana on-line”). El Proyecto Genoma Humano: una proyección hacia el futuro Los progresivos hallazgos que se van derivando del PGH, encaminados hacia una secuenciación plena de nuestro genoma, no deben conducir a un reduccionismo. Richard Lewontin lamentaba: “hace falta algo más que el ADN para constituir a un ser vivo… Una vez escuché a un destacado investigador, líder en Biología Molecular, decir en la sesión de apertura de un congreso que si tuviera un computador muy potente, y la secuencia completa del ADN de un ser vivo, podría computar al organismo. Es decir, describir totalmente su anatomía, fisiología y comportamiento. Esto es una falacia. Aunque tuviésemos la capacidad de computar toda esta información, el organismo es más que su ADN”. El PGH se plantea un desarrollo ulterior más complejo: el “proteoma”. El proteoma es el conjunto de proteínas que intervienen en los procesos biológicos de una especie. Y es que el ADN está constituido en base a sólo 4 bases nitrogenadas, mientras que las proteínas están constituidas en base a 20 aminoácidos diferentes. Además, hay que establecer la estructura tridimensional adecuada, pues esa estructura espacial es la que interviene decisivamente en el papel que cumple la proteína. La deducción de cada gen y su expresión servirá para conocer las alteraciones proteicas responsables de muchas patologías. Tres campos clínicos son los beneficiados directamente por los avances del PGH: - El diagnóstico. Para aquellas enfermedades con una base genética, el conocimiento de nuestro genoma permite desarrollar pruebas diagnósticas a nivel, incluso, preembrionario. Mediante la técnica del “clonaje posicional” puede localizarse el gen responsable de determinada enfermedad (Tabla-II).
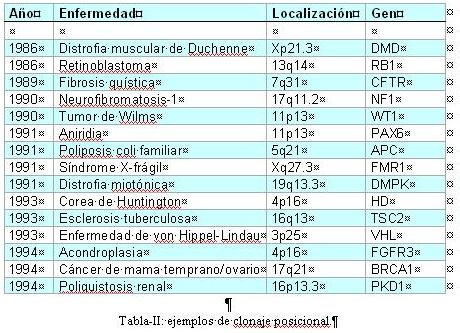 - El terapéutico. Pueden desarrollarse terapias génicas, en base a la corrección de secuencias que porten genes alterados, que posibiliten el tratamiento de determinadas enfermedades. Las células pueden modificarse genéticamente en un cultivo para, posteriormente, reinsertarlas (procedimiento “ex vivo”); pero también pueden tratarse directamente células o tejidos somáticos (procedimiento “in vivo”): Tabla-III.
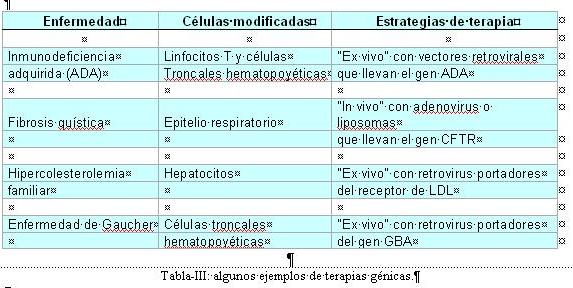 - El farmacológico. La ingeniería genética está posibilitando la producción a gran escala de proteínas con interés farmacológico: insulina, hormonas, factores de coagulación, etc. (Tabla-IV). Para ello, basta insertar -como hemos visto- las secuencias responsables en plásmidos, YACs o BACs para su clonación en organismos transgénicos (bacterias, levaduras, plantas o animales).
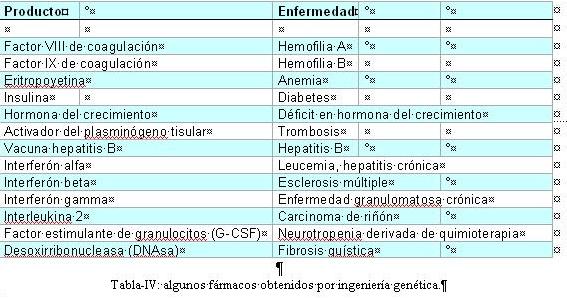 Ahora bien, los avances que el PGH y la “big-science” están brindando a la Ciencia, no están exentos de ciertos riesgos en su aplicación. La clonación de embriones o la experimentación con sus células troncales son algunos de esos potenciales peligros a los que deben hacer frente, bajo la perspectiva de la ética, biólogos y filósofos, pero también: economistas, sociólogos, médicos, teólogos, juristas,… Ante las alarmantes posibilidades abiertas en la experimentación con seres humanos, surgió una nueva disciplina: la Bioética. Aunque este tema se tratará, con más detalle, en capítulo aparte, adelantemos una breve reflexión. Por más rentable que resulte para las compańías biotecnológicas, en modo alguno resulta lícito experimentar con seres humanos, ni hacer con nuestro ADN todo tipo de quiméricas y aberrantes investigaciones. En relación con ello, cabe detallar los objetivos que, para el periodo 1998-2003, se planteaba el programa internacional ELSI. Recordemos que el “Ethics, Legal and Social Implications” figuraba entre los objetivos que el PGH se marcaba en 1988. - Examinar todo lo relativo a la complejidad de las secuencias del ADN humano y la variación genética en el hombre.
- Examinar todo lo que se refiera a la integración de las nuevas tecnologías y su utilización con fines clínicos y salud pública.
- Examinar todo lo que se refiera a la integración del conocimiento sobre genómica e interacciones de genes y medio ambiente en objetivos no clínicos.
- Explorar cómo el conocimiento genético puede inferir con una variedad de conceptos filosóficos, teológicos y perspectivas éticas.
- Explorar factores raciales, étnicos y socio-económicos que afectan al uso, comprensión e interpretación de la información genética, uso de servicios genéticos y desarrollo de normas jurídicas.
Bibliografía Anderson, Ch., 1994. NIH drops bid for gene patents. Science, 263: 909-910. Anderson, W.F., 1989. El tratamiento de las enfermedades genéticas. Mundo Científico, 6: 275-291. Anderson, W.F., 1995. Terapia génica. Investigación y Ciencia, noviembre 1995: 60-63. Arana, J., 2001. Materia, universo, vida. Ed. Tecnos, Madrid. Argüelles, J.C., 2002. El genoma humano. Un ańo después. Investigación y Ciencia, marzo 2002: 30-31. Ayala, F., 1980. Origen y evolución del hombre. Alianza Editorial, Madrid. Collins, F.S. et al, 1998. New goals for the U.S. Human Genome Project. Science, 282: 682-689. Crick, F., 1979. żHa muerto el vitalismo? Antoni Bosch, Barcelona. D’Agostino, F., 1998. Bioetica, nella prospectiva della filosofia dil diritto. Torino. Elles, R. ed., 1996. Molecular diagnosis of genetic diseases. Humana Press. Jacob, F., 1977. La lógica de lo viviente. Una historia de la herencia. Ed. Laia, Barcelona. Jouve de la Barreda, N., 1994. Clonación y manipulación de embriones humanos. Tapia, 74: 47-52. Jouve de la Barreda, N., 1995. Polémicas de la manipulación del genoma humano. Anuario del Derecho Eclesiástico del Estado, vol. X: 447-461. Jouve de la Barreda, N., 1996. Avances en Genética y su utilización en la enseńanza no universitaria. Alambique, 10: 69-79. Jouve de la Barreda, N., 2001. Genoma Umano e nuova Biología. Emmeciquadro, SEED: Scienza Educaciones e Didactica, Euresis, Milan, 13: 7-20. Jouve de la Barreda, N., 2004. Biología, vida y sociedad. Antonio Machado Libros, Madrid. 187 pp. Jouve de la Barreda, N., Gerez Kraemer, G. y Saz Díaz, J., 2003. Genoma humano y clonación: perspectivas e interrogantes sobre el hombre. Ed. Universidad de Alcalá, Madrid. 176 pp. Lacadena, J.R., 1994. żHacia una sacralización del ADN humano? Boletín de la Sociedad Espańola de Genética, 5: 5-8. Lacadena, J.R., 1998. La clonación en humanos. En: http://www.cnicemecd.es/tematicas/genetica/index.htm/ Lemoine, N.R. & Cooper, D.N. eds., 1996. Gene Therapy. Bios Scientific Publishers. Lewontin, R.C., 2001. The doctrine of DNA-Biology as ideology. Penguin Books. Melina, L., 1999. Reconocer la vida. Problemas epistemológicos de la Bioética. En: żQué es la vida? La bioética a debate. Ediciones Encuentro, Madrid. McKusic, V., 2000. Mendelian inheritance in man (catalogs of autosomal dominant, autosomas recessive and X-linked phenotypes). The Johns Hopkins Univ. Press. Miklos, G.L.G. & Rubin, G.M., 1996. The role of the genome Project in determining gene function: insights from model organisms. Cell, 86: 521-529. Monod, J., 1977. El azar y la necesidad. Ensayo sobre la filosofía natural de la biología moderna. Seix Barral, Barcelona. Olson, M.W., 1995. A time to sequence. Scienze, 270: 394-396. Sánchez-Monge, E. y Jouve, N., 1989. Genética. Ed. Omega, Barcelona. Scola, A., 1999. żQué es la vida? La bioética a debate. Ediciones Encuentro, Madrid. Vila-Coro, M.D., 1995. Introducción a la Biojurídica. Fac. Derecho, Univ. Complutense Madrid. 366 pp. Direcciones en Internet OMIM (UK): http://www.hgmp.mrc.ac.uk/omim OMIN (USA): http://www.ncbi.nim.nih.gov/Omim The National Center for Biotechnology Information: http://www.ncbi.nim.nih.gov UK Human Genome Mapping Project Resource Center: http://www.hgmpmrcac.uk/ US Human Genome Mapping Projrct: http://www.ornl.gov/hgmis/•- •-• -••••••-•
Jesús Romero-Samper
Visualiza la realidad del aborto: Baja el video

Rompe la conspiración de silencio. Difúndelo. |